

Alzheimer’s disease is a neurodegenerative condition that affects over 10% of people over 65. The observable symptoms painfully familiar: Difficulty with language, confusion and increasing memory loss. However, the molecular mechanisms underlying this disease remain mysterious.
Before the 2000’s, the only way to test if someone truly has Alzheimer’s was to examine their brain after death. This is how it was first identified in 1906 by Alois Alzheimer. He dissected the brain of a psychiatric patient named Auguste Deter and observed ‘amyloid plaque’ under the microscope.
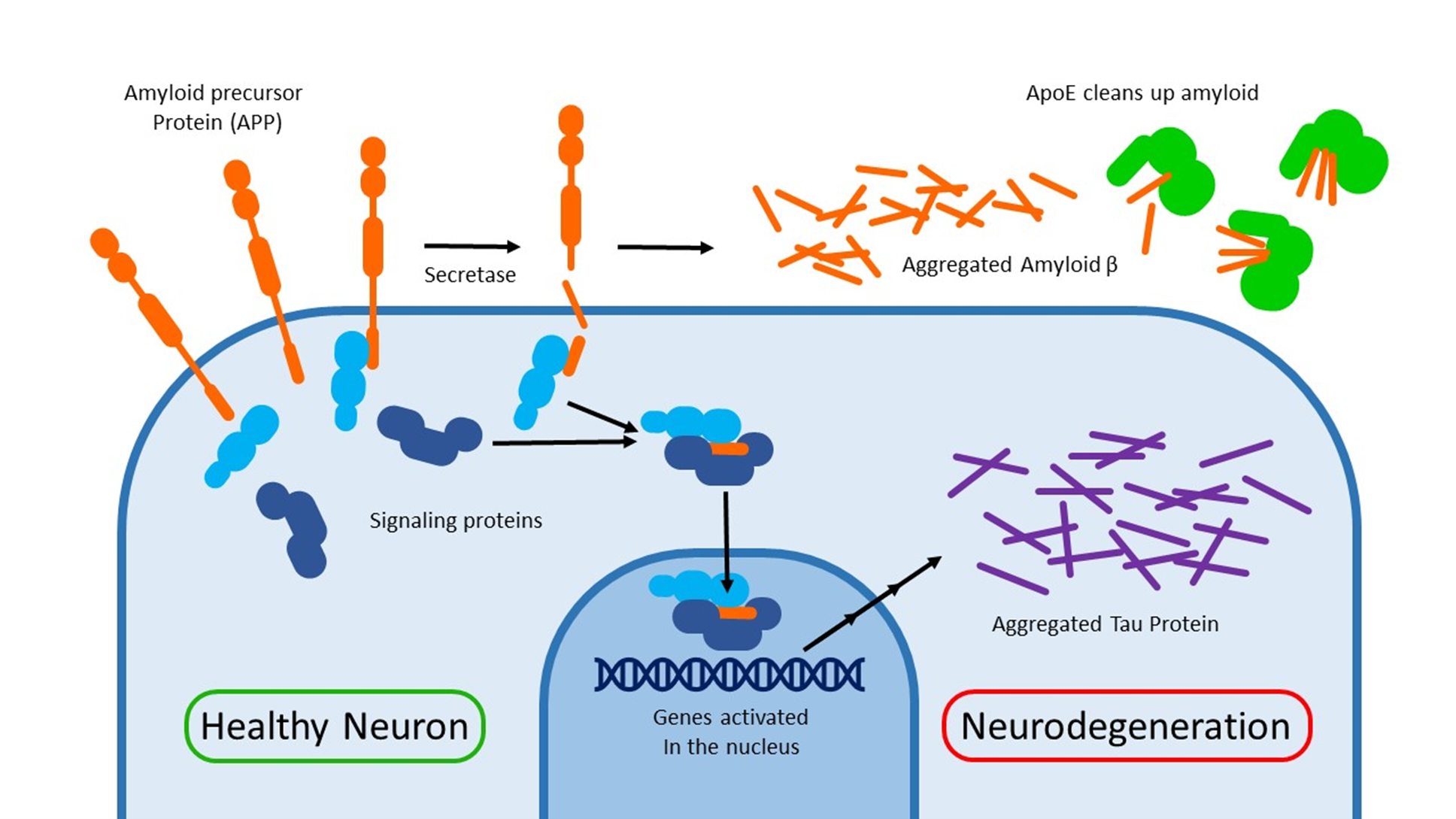
The substance that made up the amyloid plaques wasn’t determined until 1984: A tiny protein named amyloid beta. One of the reasons why this took such a long time is that amyloid beta sticks together and clogs up the protein machinery you’re using to make it!
Beta amyloid ranges from 36-42 amino acids long. Your genes determine the length and since some are more prone to aggregate than others, this contributes to the hereditary nature of the disease. Soon the gene that encoded beta amyloid was discovered. Amazingly, beta amyloid starts out as an enormous 770 amino acid precursor.
Amyloid precursor protein (APP) is expressed on the outer membrane of neurons. Its exact function is unknown. One would imagine that removing this gene could be a cure for Alzheimer’s, but ironically knocking it out of mouse models causes memory loss and impaired learning.
Many outer membrane proteins must be cut by an enzyme to achieve their function. For example: The Notch signaling protein gets cut by a secretase enzyme. Unfortunately, these cutting enzymes are rather promiscuous and sometimes react with the wrong target. APP is one of these unlucky bystanders.
When APP gets cut, the beta amyloid fragments spill out and aggregate together. Aside from getting in the way, these amyloid plaques cause inflammation and an autoimmune response. For decades, This amyloid hypothesis has been viewed as the driving force behind Alzheimer’s disease.
A key piece of evidence for the importance of beta amyloid is the genetics of another protein called ApoE. This protein helps to clear up the mess of aggregated amyloid surrounding the neurons. There are 3 major alleles of the ApoE gene throughout the population: ApoE2, E3 and E4. The last of which puts you at a greater risk for Alzheimer’s. Since you have 2 copies of your DNA (one from each parent), you can have many different combinations of APOE. 25% of people have 1 copy of E4. ~3% of people have 3 copies of E4.
The main point of controversy is this: Are amyloid plaques a cause of the neurodegeneration in Alzheimer’s, or are they just a symptom? So far, the most promising treatment strategies have only been able to slow cognitive decline but not actually cure the underlying problems.
If beta amyloid is only a symptom, what could the cause possibly be? Despite the disappointments with this tiny fragment, I feel we should not throw the baby out with the bathwater. There is so much more to the amyloid precursor protein!
The APP gene is found on chromosome 21. Trisomy 21, also known as Down’s syndrome, is a condition that causes a baby to be born with an extra copy of this chromosome. People with down syndrome are far more likely to develop Alzheimer’s disease: It begins to develop at an earlier age, and ~50% of people over 60 will suffer from it.
Some people, without Down’s syndrome, can inherit mutations in APP that make it easier for secretase to cut it. This causes symptoms to develop around age 40-50. The cleavage of APP seams to be very important, but what effect would this have other than beta amyloid production? Maybe we have the wrong end of the stick.
The other end of APP is left inside the cell after secretases cut out the beta amyloid. Binding experiments have shown that this small piece of protein interacts with signaling proteins and can even travel inside the nucleus. This alters gene expression. Some of these genes (such ask GSK3-3b) can cause another type of protein aggregate to form: Neurofibrillary tangles.
A lot of research has been focused on designing drugs to clean up amyloid. Unfortunately not a single one has been successful yet. In September 2022, a beta amyloid binding antibody drug called Lecanemab was found to be successful in phase III clinical trials. This means there is a chance this avenue could prove to be fruitful.
Image credit: Josh Riemer
{kind=link}